Module 3: OMArk
OMArk assesses the quality of protein-coding gene repertoires. It compares an extant species’ proteome to the expected gene repertoire of the lineage’s common ancestor, inferred from HOGs in the OMA database. It uses OMAmer to quickly place each query protein to its matching HOG (ancestral protein).
OMArk provides two key measures:
-
Completeness = how many of the conserved genes expected for the lineage are present.
-
Consistency = whether the proteome looks taxonomically and structurally coherent (few fragments, few contaminants, not too many “unknowns”, i.e. proteins with no detected homology).
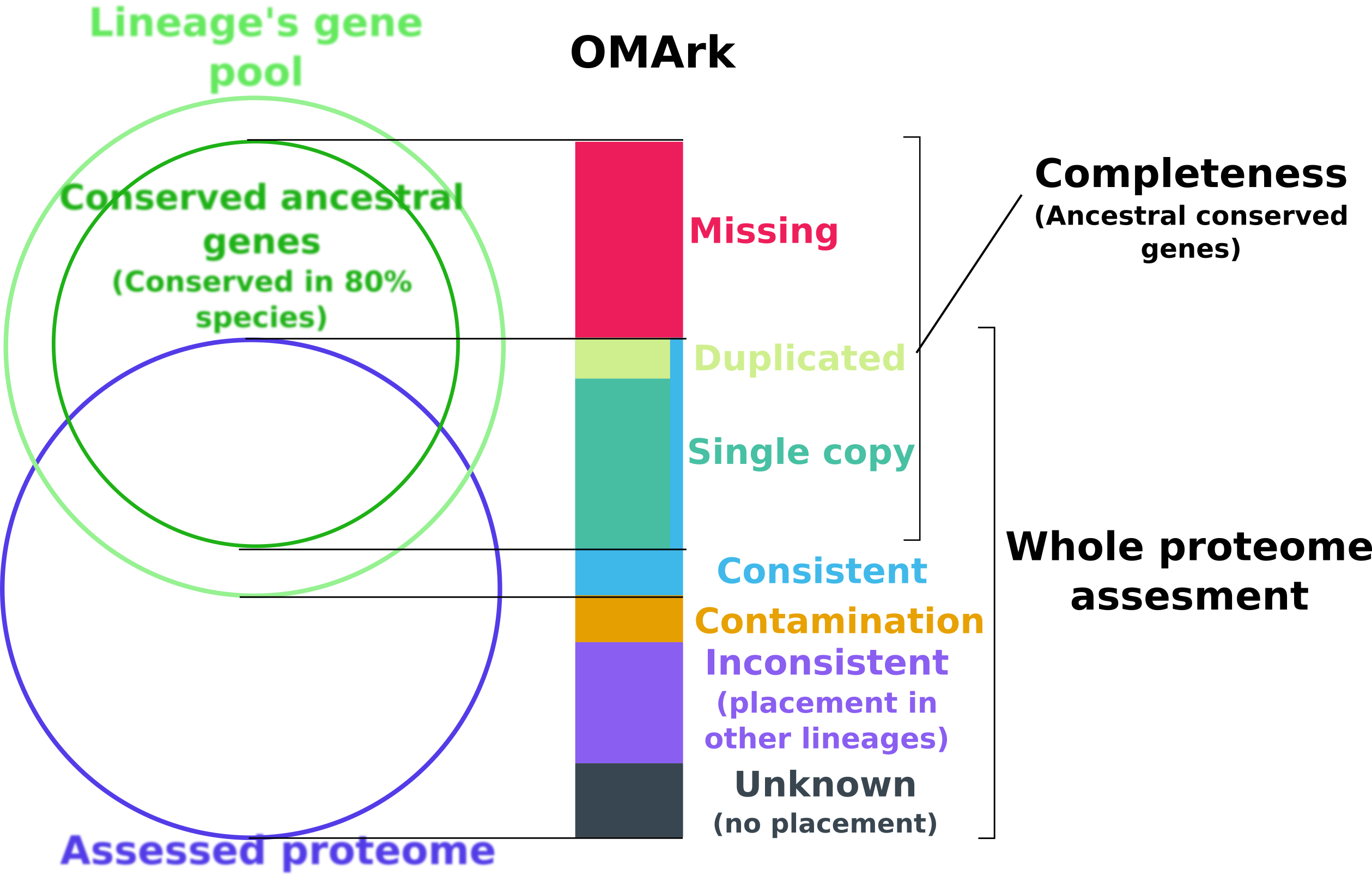
Link to the OMArk paper: Nevers et al. 2025, Nat Biotechnol.
OMArk is available as both a command-line tool (recommended for large projects) and as a web server: https://omark.omabrowser.org/ (recommended for assessing a few proteomes).
Today’s exercises: explore precomputed results in the web interface, compare species within a clade, and interpret outliers.
3.1 Clade overview
You are interested in doing a comparative genomics study of whales and dolphins. Go to the OMArk browser and navigate to the Cetacea clade using Select Taxon. Make sure you are viewing the 2024 release by clicking Change datasets.
Which three species have outlier proteome sizes?
Sousa chinensis, Pontoporia blainvillei and Eschrichtius robustus all have too few proteins compared to the other species.
3.2 Completeness assessment
Inspect completeness bars for these three outlier species.
What proportion of genes are missing?
S. chinensis ~59% missing, E. robustus ~38% missing, and P. blainvillei ~76% missing compared to ~10% or less in most other cetaceans.
3.3 Consistency assessment
Compare the stacked bars in the whole proteome assessment for the outlier proteomes vs. normal cetaceans.
What unusual patterns do you see?
S. chinensis has many “unknown” proteins; E. robustus and P. blainvillei have many fragmented proteins.How are fragments defined in OMArk?
Go to the Help section of the website.Fragments are those proteins whose length is less than half the median protein length of its gene family.
3.4 Proteome source comparison
Examine the three versions of the Narwhal (Monodon monoceros) proteomes (UniProt vs. Ensembl vs. NCBI) in the 2024.06 OMArk server release.
Which would you choose for downstream analysis, and why?
One should not choose the UniProt version of the proteome, as it has a significantly higher proportion of missing genes compared to the other two. The Ensembl and NCBI proteomes are of comparable quality. However, NCBI has the least number of missing genes, the least % of fragmented/partial hit genes, and the least number of unknown genes.
3.5 Contamination
Open the Physeter catodon proteome from Ensembl.
How many contaminants are reported and which type of organisms?
71 proteins which belong to the Sarcocystidae clade (a parasitic Apicomplexa).